6 The Science; Why We Must Care About Dose, Pharmacokinetics, Pharmacodynamics And Utility
At the end of this chapter, the reader will understand:
The basic concepts and interrelationships between dose, Pharmacokinetics (PK), Pharmacodynamics (PD) and Utility.
Why inter-individual variability (IIV) in PK and PD is the primary reason why individuals need different doses.
That IIV in PK yields a range of exposures (concentrations) and IIV in PD yields a range of responses (across individuals receiving the same dose).
For every endpoint, each patient has their own individual D-E-R relationships.
That dose is just a very crude mechanism to attempt to deliver sufficient drug to the site of action to illicit the desired PD responses.
Why we should always expect to need to change the dose to achieve the best responses in all patients.
Drugs that are suitable for “one size fits all” dosing are like diamonds; they are very rare.
For motor racing drivers, they want a car that gives them the best outcome; they win the race. To enable this, the driver needs teams of expert engineers to design, develop, build, and optimise the car. The fastest car will involve multiple experts (aerodynamicists, material scientists, engine designers etc.) to continually optimise their “product” (the car) through the use of well design experiments and modelling and simulation. In drug development, the patient is our driver; they rightly just care about their outcomes. However to use our drug as well as possible, we need drug developers and regulators who have the same “engineering” mindset to use science and well controlled experiments (trials) to make our “product” as good as it can be. Determining the best way to dose each and every patient is, like a motor racing car, not “simple”, and we should not pretend it is. So let us consider the science.
Figure 6.1 below shows the sequence we aim to understand for all patients, the individual D-E-R relationships, and ultimately the utility (benefit-risk) for each patient.
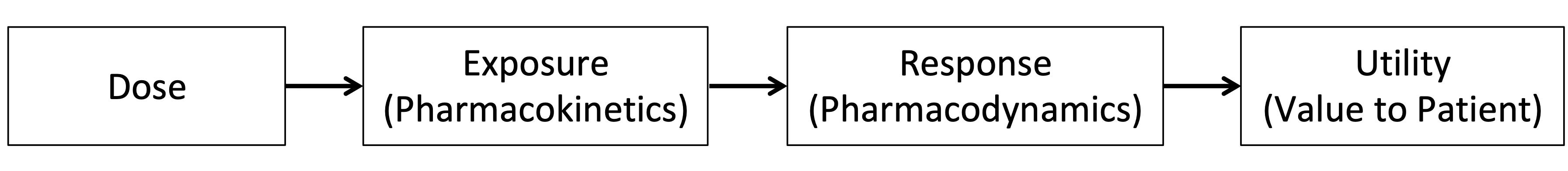
The term Exposure is used herein to broadly refer to measures of the drug concentrations achieved in the body, and the branch of pharmacology dedicated to understanding this is called pharmacokinetics.
In short:
PK can be defined as what the body does to the drug.
PD can be defined as what the drug does to the body.
Utility can be defined as the overall evaluation the patient assigns (collectively) to their individual responses.
There are numerous excellent textbooks and resources for both PK and PD, but herein we will briefly cover the essential details that need to be appreciated if we wish to get the best outcomes for patients through informed clinical trial designs.
A central component of PK is concerned with the absorption, distribution, metabolism and elimination of the drug (ADME). These play a central role in determining the blood plasma concentrations of the drug observed over time. These drug concentrations may be summarised at a particular time point (e.g. Ctrough, a “trough” drug concentration measured just prior to successive dosing events), or as the maximum drug concentration (Cmax) or as an average drug concentration (Cave) over the intra-dosing interval. Key concepts in PK include accumulation (when drug concentrations increase over time with successive doses), steady state (accumulation ends, and the drug concentrations reach a dynamic equilibrium with successive doses), and dose-proportionality/non-linearity (whether or not the magnitude of the concentrations change directly with the magnitude of the changes in dose). These basic concepts are shown in Figure 6.2 below.
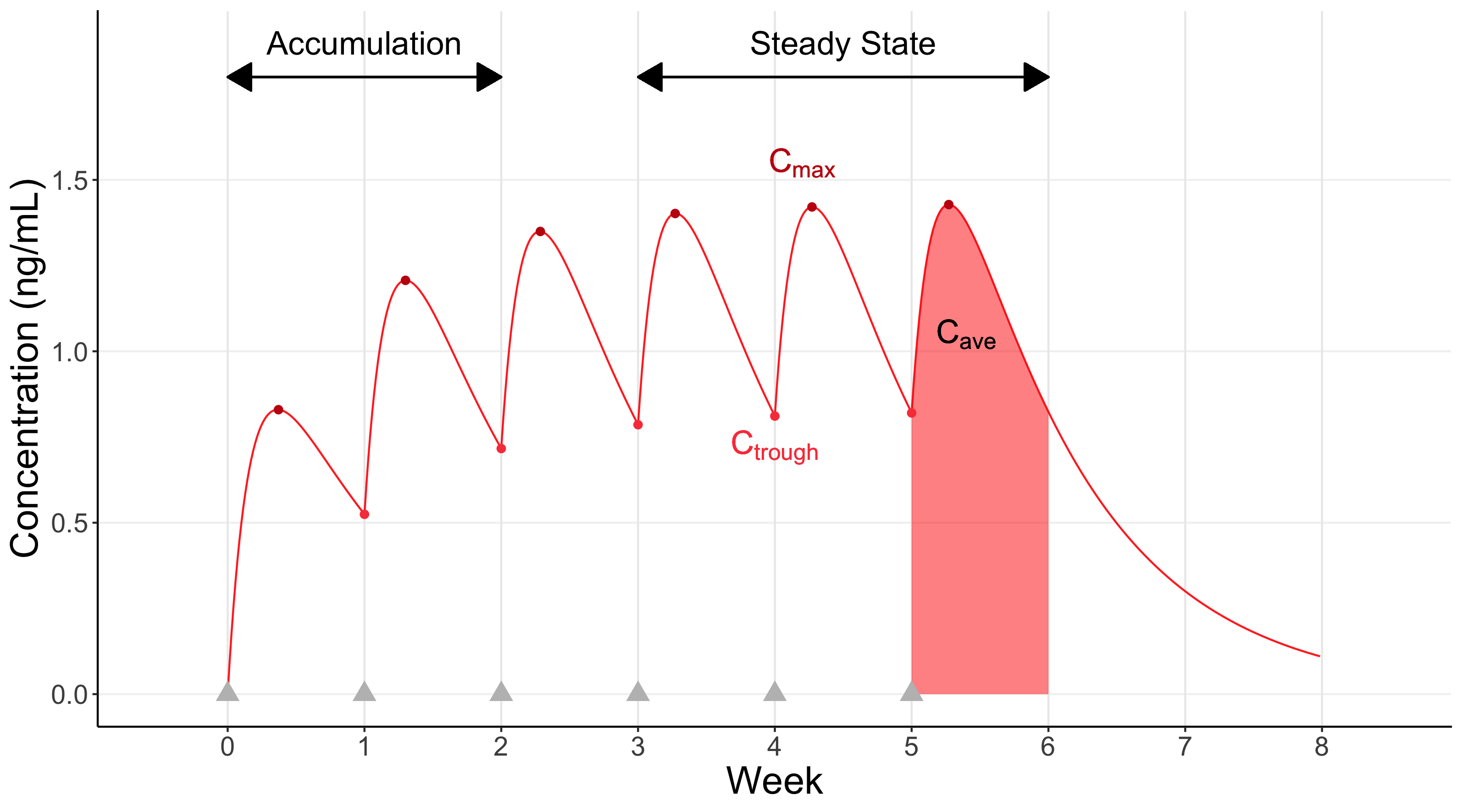
Here the individual receives weekly dosing for 6 weeks (gray triangles); steady state is achieved after around 3 weeks. Representative values for Cmax and Ctrough are highlighted, along with a visual representation of Cave, the average “area under the curve”, after the last dose (Cave will always be between Cmax and Ctrough).
In general, blood plasma drug concentrations are very important, as this will be the key pathway for the drug to reach the site of action (exceptions are typically due to the route of administration, such as inhaled drugs (e.g. asthma) or topical drugs (e.g. psoriasis)). Additional important PK considerations include drug-drug interactions (whereby taking two drugs together changes the PK of one or both drugs) and the role of active metabolites (when the (parent) drug is metabolised into a molecule that itself can induce a PD response).
PD is concerned with the biologic effects of the drug/dose/concentration, and hence are the responses that matter to the patient. These responses are often labelled as benefits (i.e. efficacy) and harms (i.e. safety/tolerability), and thus include all clinical endpoints that we may measure in a clinical trial.
Although there are cases where the PD effects are observed to change almost instantaneously with the changes in PK (a “direct” effect), it is much more common that PD changes are delayed relative to the PK concentration (an “indirect” effect). For example, the skin of a patient with psoriasis will not immediately change following their first dose, but rather will improve more slowly over the comings days/weeks/months.
The field of PK/PD modelling has developed an extensive range of flexible models to describe simultaneously how both the PK and PD change over time, with the PD changes over time being driven by the PK changes over time (with suitable delayed effects where necessary). In cases where the delay in response is substantial (e.g. weeks or months) relative to the dosing regimen frequency (e.g. daily), it is often reasonable to relate a simple measure of drug exposure, such as the average concentration (Cave), to the PD endpoint, since the fluctuations in concentrations between the dosing events (e.g. within the day) are unlikely to add further clarity beyond that provided by the simpler drug exposure measure. Notable exceptions would be when particularly high concentrations may be more strongly linked to an acute toxicity than the average concentration, in which case Cmax may be more informative that Cave. The usefulness of Ctrough is primarily in its simplicity, since it requires only a single PK sample to be taken (unlike both Cmax and Cave). In cases where there is a very strong correlation between Ctrough, Cave and Cmax, it may be acceptable to rely on a single PK sample, but generally it is much wiser to collect multiple PK samples from each patient in the drug development program, since this will allow their full PK profile over time to be predicted, and hence allow a much more accurately understand the interrelationship between the PD effects observed and the PK effects that are driving them.
Utility is a more nuanced topic that will be discussed in more detail in later chapters, but essentially is concerned with how patients actually value the trade-off between the benefits and harms. For example, if an epileptic patient achieved a 50% reduction in seizures with their initial dosing regimen, but experienced 1-4 moderately intensive headaches each month thereafter, would they consider this trade-off worthwhile (a positive utility) or not (a negative utility)? In addition, would they prefer to explore lower/higher doses to find a better dose for them (one with a higher utility)? For the remainder of this chapter, the role of utility will be temporary paused, and we will focus on the first three components, the sequence from dose to PK to PD across our heterogeneous patients.
As Woodcock [1] wisely wrote:
“The principal challenge in therapeutics is the variability of human responses to drugs, both for good and for ill.”
If we wish to intelligently dose drugs, I would posit that the most important aspect to understand and account for is the inter-individual variability (IIV) in both PK and PD across patients.
That is:
For every endpoint, each patient has their own individual Dose-Exposure-Response relationships.
This means that every patient will have their own individual Dose-Exposure relationship (IIV in PK) and their own individual Exposure-Response relationship (IIV in PD). Thus any fixed-dose regimen given to a group of patients will yield a wide range of exposures (drug concentrations), and the responses to these drug concentrations will also differ between patients. Figure 6.3 shows one way we can illustrate the relationship between Dose, Exposure and Response for 6 hypothetical patients depending on whether we have a “Fixed Dose”, “Fixed Exposure” or “Fixed Response” (these are essentially selected points on the individual D-E-R curves for each of the 6 patients).
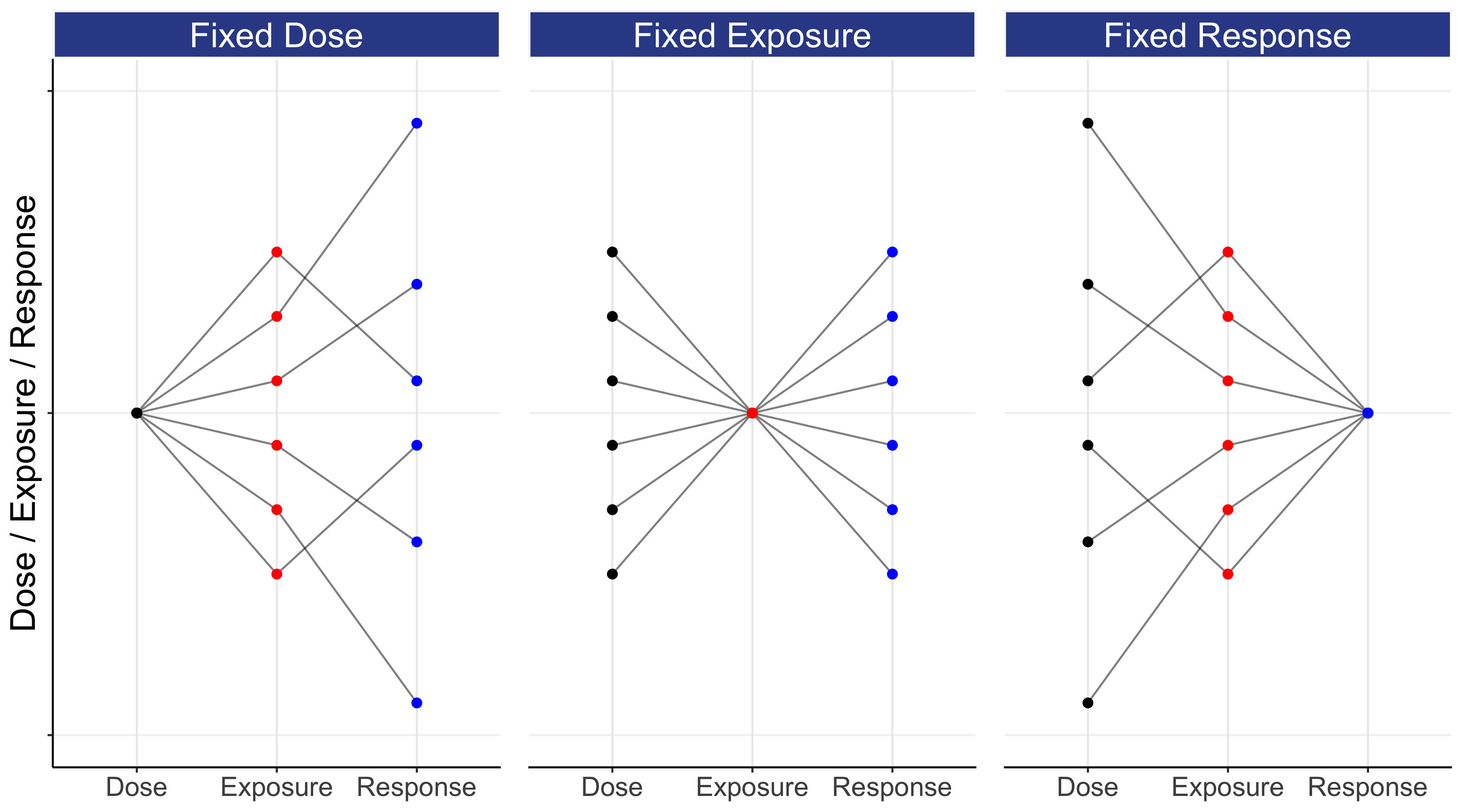
This figure may take a moment to fully grasp, since the y-axis is simultaneously quantifying Dose, Exposure and Response. In the left panel, all patients receive a “Fixed Dose”, and this leads to a wide range of exposures (e.g. Cave), that in turn lead to a very wide range of responses. In the middle panel, all patients now have the same exposure (for example, the dose has been titrated to achieve a given exposure). Here we see that a wide range of doses are needed to achieve the same exposure (due to IIV in PK), and that the same exposure leads to a wide range of responses (due to IIV in PD). In the right panel, we have the same responses for all patients (for example, the dose has been titrated to effect, either with or without the use of exposure measures). Note the cumulative effect of the variability as we travel from either dose to response (left panel), or from response to dose (right panel); stately alternatively, giving the same dose to all patients will lead to a wide range of individual responses, whilst obtaining the same response in all patients will require a wide range of individualized doses. Thus although giving the same dose to all patients is “simple”, it is not what we seek; we seek to get the same (good) response in all patients.
Although clearly an oversimplification of the complex cascade from dose to response, the above illustrates a very important point:
Dose is just a very crude mechanism by which we seek to deliver sufficient drug to the site of action to illicit the desired PD responses.
Thus we should always expect to need to change the dose to achieve the best responses in all patients; we need Personalised Dosing because of the omnipresence of IIV in PK and PD.
It is useful to contrast how each panel in the figure above translates to a drug development strategy.
Fixed Dose: What is the “optimal” one-size-fits-all dose.
Fixed Exposure: What is the “optimal” target exposure (e.g. Therapeutic Drug Monitoring)
Fixed Response: What is the “optimal” dose-titration algorithm.
As concrete examples, we can compare two drugs used to treat type 2 diabetes. The dipeptidyl peptidase-4 (DPP-4) inhibitor sitagliptin (brand name Januvia) is given to all patients at a 100 mg dose (“Fixed Dose”). The long acting insulin, insulin glargine (brand name Lantus), is titrated to effect using self-measured blood glucose levels (“Fixed Response”). With lifetime sales of >$30 billion dollars (sitagliptin) and >$60 billion dollars (insulin glargine), both drugs have been commercially very successful. Based on the pharmacology and mechanism of action (MoA) of each drug, both drugs were sensibly developed and commercialised. These drugs will be discussed furthermore in this book, but briefly 100 mg of sitagliptin is a very high dose, since this dose is approximately 20 times the ED50 (the dose which elicits 50% of the maximum drug effect) [2]. Importantly, DPP-4 inhibitors are remarkably “clean” drugs with excellent safety/tolerability profiles, allowing such a very high dose to be given to all patients. If patients fail to response adequately to 100 mg, it would be unwise to consider an even higher dose; rather, the patient just does not response well to drugs with this MoA. In contrast, too high a dose of insulin glargine can lead to hypoglycemia (very low blood glucose) and hence it is sensible that the patient slowly up titrates from an initial low dose. Insulin glargine is delivered subcutaneously using injection pens where the patient turns a dial on the pen to select the right dose (a brilliantly simple and effective dosing device). The ability for patients to tailor their insulin glargine dose to achieve the desired response is the key to the success of insulin glargine; it has been very successful because the dose can be titrated appropriately, and not despite the fact that the dose needs to be titrated.
Drugs like sitagliptin are very rare, and indeed we will refer to types of drugs herein as diamonds; they exist, but are scarce among our much more common coals. Most drugs are coals; they do require us to carefully balance the benefits and harms at the patient level.