5 Dose-Response Trials; A Brief History And Overview Of Current Practices
At the end of this chapter, the reader will understand:
How the 1994 ICH E4 guideline “Dose-Response Information to Support Drug Registration” has shaped modern phase 2 D-R trial designs.
How ICH E4 rightly distinguishes between Population (average) D-E-R relationships determined from Parallel, fixed-dose, D-R trials and Individual D-E-R relationships determined from Individual Titration D-R trials.
How the simplicity of the analysis, and not optimizing individual patient outcomes, led ICH E4 and the FDA to wrongly promote Parallel, fixed-dose, D-R trials over Individual Titration D-R trials (since the latter required “careful analysis”!).
How most modern phase 2 D-R trials are poorly designed, poorly analysed, or both.
If we wish to refocus drug development to use dose to maximise individual patient outcomes, we need Individual Titration D-R trials that can determine Individual D-R relationships.
On the topic of how to design D-E-R trials so that we learn how best to dose each and every patient, we will start with a (mis) quote from the journalist H L Mencken:
“For every complex problem there is an answer that is clear, simple, and wrong”
Most of the designs and analyses of D-E-R trials are indeed clear, simple, and wrong. Let me explain why.
To understand modern D-E-R trials, two important events/observations from the past need to be discussed. Firstly the critical role played by the 1994 ICH E4 guideline “Dose Response Information to Support Drug Registration” [1]. Although this document in nearly 30 years old, the authors made many sensible observations; a few are included and discussed below.
On the issue of selecting a starting dose:
“What is most helpful in choosing the starting dose of a drug is knowing the shape and location of the population (group) average dose-response curve for both desirable and undesirable effects. Selection of dose is best based on that information, together with a judgement about the relative importance of desirable and undesirable effects.”
Here the authors recognized that the judicious selection of the starting (initial) dose can be guided by understanding the shape of the (population) D-E-R for both efficacy and safety/tolerability (without this information, there is indeed no scientific basis for the starting/initial dose).
On the issue of how to titrate the dose for a patient:
“In adjusting the dose in an individual patient after observing the response to an initial dose, what would be most helpful is knowledge of the shape of individual dose-response curves, which is usually not the same as the population (group) average dose-response curve. Study designs that allow estimation of individual dose-response curves could therefore be useful in guiding titration, although experience with such designs and their analysis is very limited.”
This astutely acknowledges that patients follow their own individual D-R relationship, and that this is different to the population D-R relationship.
On the issue of the design of parallel, fixed-dose, D-R trials:
“The parallel dose-response study gives group mean (population-average) dose responses, not the distribution or shape of individual dose-response curves.”
“It is all too common to discover, at the end of a parallel dose-response study, that all doses were too high (on the plateau of the dose-response curve), or that doses did not go high enough. A formally planned interim analysis (or other multi-stage design) might detect such a problem and allow study of the proper dose range.”
Thus the authors understood that the outcomes from such trials only yield the population (average) D-R; we do not learn individual D-R relationships. In addition, they mention the value of an interim analysis/adaptive design to ensure the right dose range is actually explored in such trials (discussed further in the chapter on adaptive randomisation, see Chapter 19).
Under the heading “Parallel dose-response” the guideline states (with my emphasis in bold):
“Randomization to several fixed dose groups (the randomized parallel dose-response study) is simple in concept and is a design that has had extensive use and considerable success.”
Under the ominous heading of “Problems with Titration Designs” perhaps the most relevant sentence to explain why we are where we are today is the following (with my emphasis in bold):
“A study design widely used to demonstrate effectiveness utilizes dose titration to some effectiveness or safety endpoint. Such titration designs, without careful analysis, are usually not informative about dose-response relationships. In many studies there is a tendency to spontaneous improvement over time that is not easily distinguishable from an increased response to higher doses or cumulative drug exposure. This leads to a tendency to choose, as a recommended dose, the highest dose used in such studies that was reasonably well tolerated.”
In 2018 a highly experienced regulator, Robert Temple (Deputy Center Director for Clinical Science at the FDA), gave a presentation on the Design of Clinical Trials.
It included this text regarding D-R:
“Goal: Define D/R curve for benefits and risks
Until early 1980’s, most trials with more than one dose titrated the dose, generally to some endpoint. This meant:
The group on any given dose was not chosen randomly
Time and dose were confounded; secular trend would look like response to dose. Particularly useless for safety
In 1980’s, FDA promoted the randomized, parallel, fixed dose, dose-response study, identified as the standard in ICH E4 guidance.”
Thus the above texts provide us with an explanation of why parallel, fixed-dose designs for D-R trials where (incorrectly) deemed preferable to titration based designs (for brevity, these will be referred to a Parallel and Individual Titration below).
Parallel, fixed-dose designs for D-R trials were promoted as superior to Individual Titration trials because they are just “simpler” to analyse. However they are not best for patients!
Both the ICH E4 text and that from Robert Temple rightly state that a simple analysis of titration trials that ignores the (partial) confounding of dose and time would be flawed; however this absolute does not mean they titration trials are unanalyzable – it just means exactly what ICH E4 states, a “simple analysis” does not suffice! Indeed, the difference between fixed-dose regimens and placebo in a Parallel trial also changes as a function of time (duration of treatment), but no one would suggest we cannot describe/model how the dose effects evolve over time (relative to the placebo response, which also changes with time!). Furthermore, whilst the dose may remain the same over time, the drug concentrations achieved with repeated doses typically increase over time, so a “fixed-dose input” is “fixed” in name only, as the system (the patient) actually experiences a “changing concentration input” over time with repeat dosing (if this point is unclear, later chapters will introduce the key PK principles that underpin and explain this observation).
In addition, there are both errors and significant omissions in ICH E4. Under “Guidance and Advice”, it states:
“A widely used, successful and acceptable design, but not the only study design for obtaining population average dose-response data, is the parallel, randomized dose response study with three or more dosage levels, one of which may be zero (placebo). From such a trial, if dose levels are well chosen, the relationship of drug dosage, or drug concentration, to clinical beneficial or undesirable effects can be defined.”
The above suggests that only 3 dose levels (such as placebo, 10 mg and 20 mg) would be a “successful and acceptable design” to define both benefits and harms. This is incorrect. To fit an appropriate D-R model, you need a minimum of 4 dose levels, and these need to be well spaced over the “right” part of the D-R relationship (see Chapter xxx). To be clear, a design such as Placebo, 10 mg and 20 mg is incapable of adequately describing any D-R relationship.
ICH E4 also has a glaring omission; it fails to suggest how to actually “link” doses together using a suitable D-R model(s). No D-R models are discussed or considered; the actual analysis of D-R trials is wholly absent. How can any D-R relationship be determined without a D-R model? Clearly one can “join the dots” of the mean responses at each dose (like in the Placebo, 10 mg, and 20 mg example), but this is an awful and painfully unscientific strategy (this will be illustrated later in Chapter 11). Since ICH E4 is still an active guideline, I feel it is important to shine a light on these major errors and omissions. Good D-R trial design requires the investigation of well-spaced dose levels over a wide dose range in sufficiently large sample sizes. The analysis then combined all data together using a suitable D-R model. Thus we need both the right design and the right analysis, and that requires a D-R model!
Finally, it is important to note that the criticism of parallel group dose ranging trials herein is not new. In a 1989 article entitled “Study Designs for Dose Ranging”, Sheiner, Beale and Sambol [2] wrote:
“We believe one must begin with a parametric model for patient-specific dose-response curves. Knowledge of the distribution of these curves in a population provides a basis for choice of an initial dose (e.g., the dose that achieves a given response in a given fraction of patients) and, after observation of response to an initial dose, for choice of an incremental dose for a specific patient (by use of Bayes rule). The current parallel-dose design can provide only poor information about the distribution of dose-response curves, biased estimates of the typical curve, and little information on interpatient variability”
In their discussion, they added:
“The dose-escalation design imitates clinical practice and was a popular design for dose-ranging studies in the United States (it continues to be so in Europe) until replaced by the parallel-dose design. It is instructive to examine why the latter appeared preferable. In brief, we believe design and analysis flaws, which are obviated to a large extent by the approach described in this article, were responsible. Apparently, rather than analyze and correct the flaws, researchers chose to abandon the design. In our opinion, this was a case of throwing out the baby with the bathwater.”
I fully agree.
Returning to our primary comparison around Parallel and Individual Titration designs, we need to be very precise in the comparison between these two D-R designs.
Parallel and Individual Titration D-R designs are different, the data acquired is different, the analysis is different, and the focus of inference is different (population D-R or individual DR). The questions they address have both similarities and differences.
At the highest and simplest level we can compare the two designs based on the final outcomes they achieve (versus placebo or an active control). For example:
Parallel - if all patients receive the same fixed-dose regimen, what are their outcomes after 6 months?
Individual Titration - if all patients use the same dose titration algorithm, what are their outcomes after 6 months?
Thus we see our first similarity. Both designs generate a set of individual patient outcomes (for both efficacy and safety) that can easily be compared. However with Parallel, we are forced to maintain the exact same dose throughout, irrespective of how well or poor the intermediate outcomes are for each patient; despite on-treatment data/evidence, we deliberately aim to not change the dose! As such, I see such fixed-dose regimens as appealing to fatalism – we are (apparently) resigned to the fact that the benefits and harms caused by the dose to the patient are unavoidable and inevitable; the patient must experience the full frequency and severity of tolerability/safety effects that has been assigned to them; this is not OK. Our need for “simple” data is being placed above our need for better patient outcomes; this is wrong. With Individual Titration, we are guided to change the dose to maximize the benefits and minimize the harms caused by the dose for each patient. In contrast to the Parallel patients, the dose for each patient may be adjusted based on accruing observed intermediate outcomes, and hence we have the opportunity to use dose to improve their final outcomes.
In a Drug Development For Patients world, if we have the opportunity to improve individual patient outcomes by changing their dose, are we not compelled to do so? I strongly believe we are. Do you?
For well designed Parallel D-R trials, we get the necessary information to construct the Population D-R that, in some cases, will be reasonable. For example, we may get 100 patients for placebo and 5 different dose levels (600 patients in total). Thus the resulting D-R modelling will determine the population (average) effect at each dose under the presumption that we will give all patients that dose (i.e. with no titration). For Individual Titration, we would get 100 placebo patients and 500 individual D-R curves. Depending on the design used (e.g. forced titration), patients will generate data across their own dose range, and the resulting D-R modelling would determine Individual D-R relationships. Clearly care needs to be taken in both the design and analysis of these trials, but rest assured, technically we can do this!
It is worth stressing that population D-R effects are only accurate if we specifically prohibit any dose titration (from a “do no harm” perspective, why would we want to prohibit dose titrations?). For example, if a population D-R analysis determined that a dose of 100 mg would result in an average heart rate increase of 10 bpm, we may not expect the same average 10 bpm increase at 100 mg if this dose was towards the higher end of a dose titration range (e.g. 10-100 mg). This is because some patients (e.g. those with higher than average drug concentrations and/or more sensitive to the drug) may be adequately treated at lower doses, and hence never reach 100 mg. Thus from a titration perspective, we would be more interested in the heart rate effect at 100 mg only for those patients who are actually titrated from, say, 50 mg to 100 mg (that is, the cohort of patients who were inadequately treated at 50 mg). Population D-R does not answer this important question; rather it only ever addressed the “one-size-fits-all” dose effect when any form of titration is specifically prohibited.
With Parallel D-R trials, we answer a simple question, but in doing so place a paralyzing inability to adjust the dose for the patient, something we know we need to do! For anesthetists it would also be “simpler” to initiate the infusions/flow of anesthetic agents and then go home, but there is a good reason why they do not. They care about, and seek to optimize, individual patient outcomes.
When we weigh up Parallel versus Individual Titration D-R trials, we may ask “Do we want an easy design/analysis that answers a less important question, or a design/analysis that can answer the right question?”
In his book entitled “Dose Finding in Drug Development”, the author Naitee Ting wrote:
“There are some advantages of a titration design. For example, a study with this design will allow a patient to be treated at the optimum dose for the patient; this dose allocation feature reflects the actual medical practice. However, the disadvantage of a titration design is the difficulty in data analysis.”
I think this “screams” the choice we must make; should we choose the path that allows each patient to be treated at their optimal dose, or should we choose the path that avoids a difficult analysis? Do we care more about having a simple analysis than our patients?! Perhaps to state the problem more pointedly, should we allow a patient with cancer to die on their fixed-dose regimen, because we cannot be bothered to monitor markers of efficacy (e.g. tumour growth) and hence consider a dose change for them? If your partner/child were in such a trial, what design would you choose, and why?
In summary, the above text has sought to illustrate why Parallel group D-R trials were originally seen by some in the 1980s/1990s as superior to Individual Titration D-R trials, however the primary motivation was based on the simplicity of the analysis, and not on obtaining the best patient outcomes. Thus although ICH E4 is still sound in many regards, it is also now painfully outdated and desperately needs updating. It fails to show the significant weaknesses with Parallel D-R trials, and the real value to patients of Individual Titration D-R trials. If our goal is to have simple trials and “one-size-fits-all” doses, then Parallel is OK. If our goal is to obtain the best outcome for each and every patient via informed Personalised Dosing, we need well designed Individual Titration D-R trials.
Given that above history, it is perhaps not surprising to know that the current landscape of phase 2 dose-ranging trials is dominated by Parallel, fixed-dose, D-R trial designs. Unfortunately not only are these designs ubiquitous, the standard of their designs and analyses are frequently very poor.
To illustrate this point, I searched “Clintrials.org” for completed phase 2 trials with the statistical analysis plan available (so I could find the more technical information). Table 5.1 below shows some details of the first 5 trials I found (to be more representative of the industry as a whole, I did select trials from larger pharma companies, since you may expect these companies to have sufficient resources to design these trials well).
These trials were generally designed in the last 5-7 years, and look representative of the (weak) designs and analysis of “dose-ranging” trials I often see.
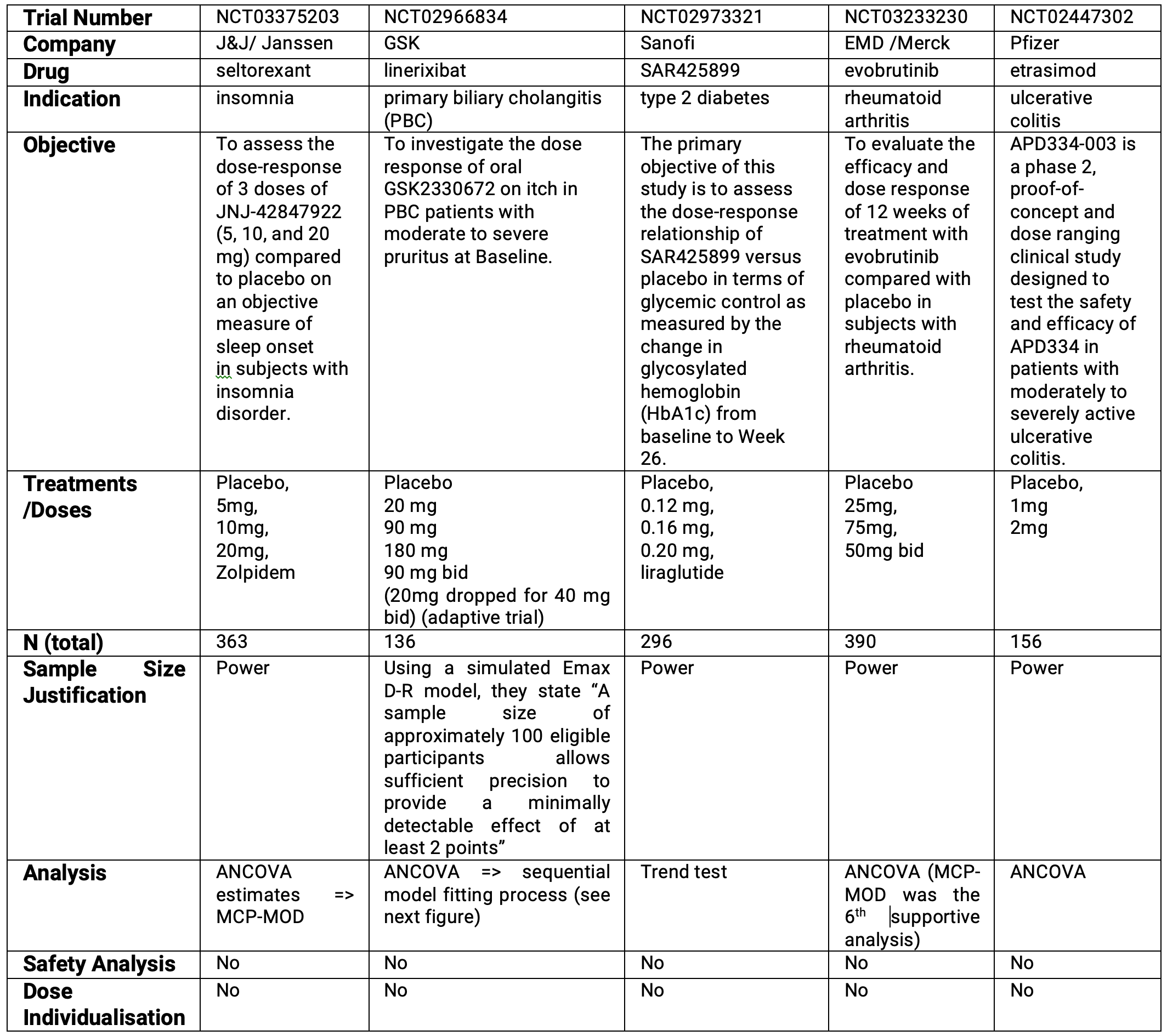
Relative to the objective, to understand the dose-response relationship, these trials are universally weak. The designs have very few dose levels across very narrow doses ranges (4/5 trials). The sample sizes are based on power statements (4/5 trials), which means they are justifying the sample size on the ability of the design to reject the null hypothesis of “no drug effect”. This is completely different to understanding the dose-response! Being able to conclude at the end of the trial that “the drug does something”, is not the same as being able to accurately and precisely quantify the D-R; the latter requires appropriate designs and greater sample sizes. The analysis methods are poor, either based on pairwise comparisons (i.e. ANCOVA), trend tests or the better (but still weak) MCP-MOD (more on this later). In addition, none of these trials reported any planned D-R analysis of safety/tolerability endpoints in the statistical analysis plan (although there may well be a pharmacometrics analysis plan with such details). A critical objective of drug development is to understand how safety/tolerability change as a function of the dose, yet here it is unclear if any D-R analysis of the safety/tolerability endpoints has been prospectively planned, or if the design is suitable for these crucial analyses. Without such analyses key decisions, such as phase 3 doses to consider, may be forced to rely on just the observed point estimates for each dose (the limitations of which will be discussed in later chapters). Finally, none of these trials considered dose-individualisation (i.e. Individual Titration D-R trials); all were fixed dosing regimens.
The GSK trial was less disappointing, with a 9-fold dose range, and a sample size justified based on something other than a simple power statement. However this trial came with a flow chart for the analysis. This is shown in the Figure 5.1 below, and is a clear example of poor D-R modelling.
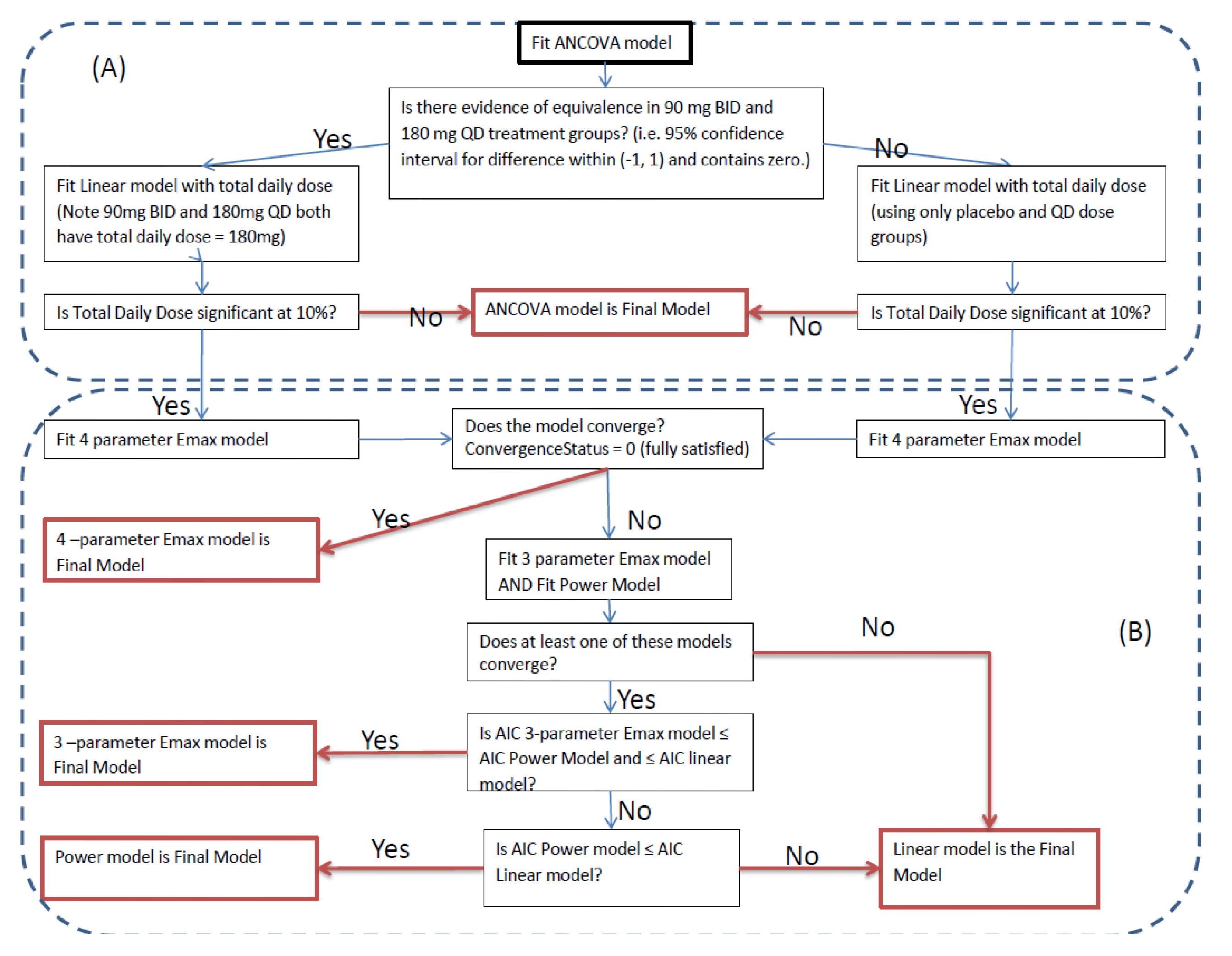
The diagram is a mix of questionable logic, significance testing and poor model selection criteria; perhaps my reaction is extreme 😀, but I see this D-R analysis being brutally butchered before my eyes! Firstly, the 90 mg bid versus 180 mg qd doses are tested as to whether we can reject the hypothesis that they are equivalent. Deciding whether to progress a once or twice daily regimen is not trivial, since the differences between these two regimens will be generally small; you need very large trials to quantity these differences. Thus this first step can only lead to pooling the two regimens (hence assuming once and twice daily are identical, which they are not) or, worse still, concluding the twice daily regimen is markedly better than the once daily regimen, which in itself would be a spurious finding (that is, you will only conclude the twice daily is significantly better in the few occasions where, by chance, the observed difference was unusually large). The analysis does then look to fit the very sensible 4 parameter (sigmoidal) Emax model, but then relies on the software to decide if this model should be accepted! If a sensible D-R model fails to “converge”, you must first ask “Why?” This will require both the data and the software fitting information output to be reviewed, and then to determine how best to proceed (perhaps the model simply needs to be reparameterised). Instead, the analysis blindly follows a sequential set of ever more naive models, potentially leaving us with the (never appropriate) linear D-R model. From an “easy of upfront programming” perspective, the flow diagram is wonderful. Unfortunately from an “accurately and precisely quantify of the D-R relationship” perspective, it is not sound.
Please understand I have no issue with any of the companies above, and the very small sample of trials above may not be a fair reflection of how any of these companies typically approach dose-response trial designs (e.g. a friend mentioned that the Pfizer trial is a phase 2A trial, with wider dose ranges studied in phase 2B). In addition, I could equally have commented on other major companies/trials, including Roche/Genentech (Fenebrutinib (NCT02833350)), Novo Nordisk (semaglutide (NCT02453711)), Abbvie (tilavonemab (NCT02880956)), Novartis (LCI699 (NCT00758524)) and Lilly (tirzepatide (NCT03131687)). All these dose-ranging trials are limited in either their design, their analysis, or both. The key point here is not to “bash” any company, but rather to show that weak D-R trials are quite common across industry, and not just limited to a few “rogue” companies.
There are also examples of much better D-R trial designs that considered very wide dose ranges (as advocated herein). For example placebo and a 40-fold dose range (Pfizer trial NCT03985293), a 60-fold dose range (Novartis trial NCT03100058), and a 16-fold dose range (GSK trial NCT00950807). This is very important, because knowing if lower doses may do as well as much higher doses for efficacy, but much better for safety/tolerability, is crucial to finding the right dose range for patients. For example in trial NCT03926611 Novartis observed similar efficacy of remibrutinib versus placebo across the dose range, from 10 mg qd to 100 mg bid (a 20-fold dose difference!). Without these lower doses, perhaps the 100 mg bid dose would have been progressed into phase 3, even though much lower doses appear similarly effective. It is also worth mentioning another excellent trial at the right end of the spectrum, one by Lilly [3]. Stage 1 of the dulaglutide phase 2/3 adaptive trial (AWARD-5 (NCT00734474)) investigated a 12-fold range of dulaglutide doses simultaneously across 2 efficacy endpoints and 2 safety endpoints using a Bayesian adaptive, D-R design. They sought to determine the two doses with maximum utility (the “trade off” between efficacy and safety) in stage 1, before selecting these doses for stage 2 (the phase 3 part of this seamless trial). Although there are many ways this trial could have been even better, it was clear that the team prospectively evaluated (via simulation) how to design the trial to combine D-R models for efficacy and safety. Stage 1 of this design is particular good, and reflective of the sound work of Donald Berry and colleagues; stage 1 is actually similar to one type of drug development strategy that will be discussed in Chapter xxx, and hence is worth reading.
In contrast to the wide dose ranges and adaptive trial designs discussed above, most oncology “dose-finding” trials are particular weak, as often only 1-2 (very high) doses are actually considered in phase 2/3. Given the myriad of serious safety/tolerability problems experienced by patients with many oncology dosing regimens, the lack of well-designed and analysed D-R trials in oncology is particularly disappointing. Simply exposing patients to very high doses is neither scientifically sound nor ethical drug development.
By the end of this book, I hope it will be clear why we generally need to study very wide dose ranges in sufficiently large sample sizes. We need our trials/programs to accurately and precisely quantify the D-R relationships for efficacy and safety/tolerability. In addition, most trials only consider fixed-dose regimens, with the expectation that a single “one-size-fits-all” dose will be appropriate for all patients. The idea that patients could achieve better outcomes with individualised dosing (i.e. personalised dosing) is often regrettably absent from these trials; I hope this will change. Remember, patients are not fields!
This section has hopefully given a short introduction to the history of D-R trial designs, and how ICH E4 has unfortunately led to Parallel, fixed-dose, D-R trials to determine Population D-R relationships being used because they are “simple”. In addition, a brief review showed that some of these trials are of a low standard. Finally, if we wish to refocus drug development to use dose to maximise individual patient outcomes, we need Individual Titration D-R trials that can determine Individual D-R relationships; Sheiner, Beale and Sambol were right 😀.